
La pharmacocinétique, ça vous semble compliqué ? C’est juste l’étude du trajet d’un médicament dans votre corps.
Ce guide décompose tout en 4 étapes simples (ADME) pour comprendre comment ça marche vraiment.
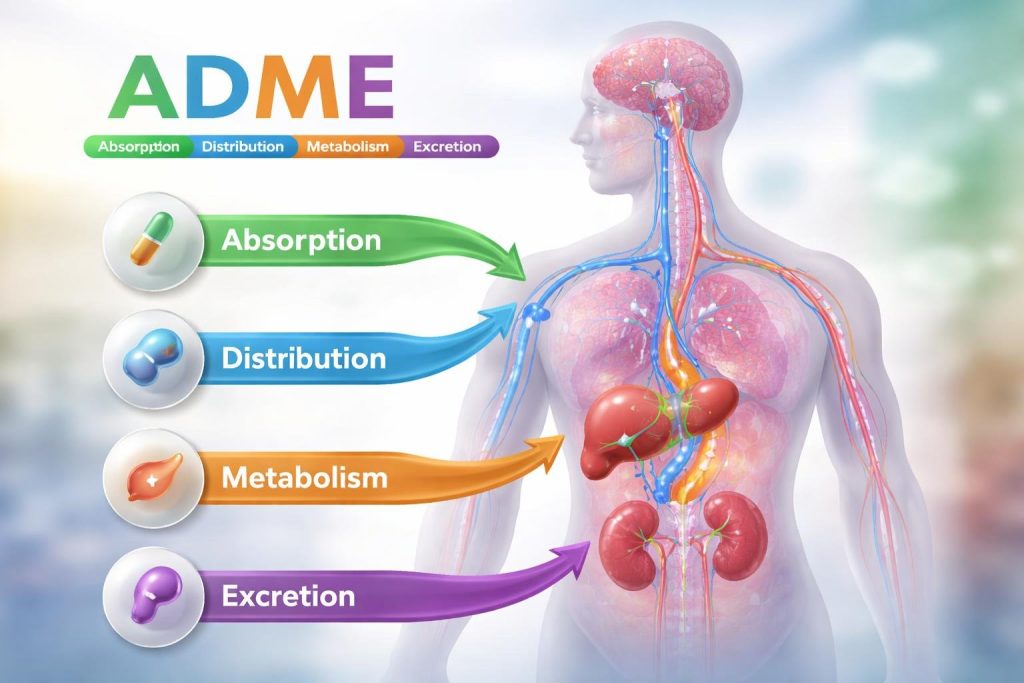
Les 4 phases de la pharmacocinétique : l’acronyme ADME
La pharmacocinétique décrit ce que l’organisme fait à un médicament. C’est l’inverse de la pharmacodynamie, qui explique ce que le médicament fait à l’organisme. Pour simplifier, on utilise l’acronyme ADME qui résume les quatre grandes étapes.
Ces étapes ne sont pas toujours séquentielles. En réalité, elles se chevauchent et se déroulent en même temps. Un médicament peut commencer à être éliminé alors que son absorption n’est pas encore terminée.
- A pour Absorption : C’est le passage du médicament depuis son lieu d’administration (bouche, peau, muscle) vers la circulation sanguine.
- D pour Distribution : Une fois dans le sang, le médicament se répartit dans tout le corps, vers les différents organes et tissus.
- M pour Métabolisme : Le corps, principalement le foie, transforme chimiquement le médicament pour le préparer à être éliminé.
- E pour Élimination (ou Excrétion) : C’est l’évacuation du médicament et de ses produits de transformation (les métabolites) hors de l’organisme.
Phase 1 : L’Absorption (A) du médicament dans la circulation
L’absorption est la première des étapes absorption distribution métabolisme élimination. C’est le point de départ du voyage du médicament. Son efficacité dépend directement de la qualité de cette étape. Pour qu’un principe actif agisse, il doit d’abord atteindre la circulation sanguine générale.
Ce processus est complexe et dépend de nombreux facteurs, liés au médicament lui-même, à la voie d’administration choisie et à votre propre corps.
Mécanismes et facteurs influençant l’absorption
Le passage du médicament à travers les barrières du corps (comme la paroi de l’intestin) se nomme la résorption. Le mécanisme le plus courant est la diffusion passive. Le médicament traverse les membranes cellulaires sans aide et sans dépenser d’énergie.
Pour que cette diffusion fonctionne, le médicament doit remplir certaines conditions :
- Être assez hydrosoluble : Il doit pouvoir se dissoudre dans les liquides du corps pour arriver jusqu’aux membranes.
- Être assez liposoluble : Il doit aimer les graisses pour traverser les membranes cellulaires, qui sont lipidiques.
- Avoir une forme neutre : Les molécules chargées électriquement (ionisées) passent très mal les membranes.
Le pH de l’environnement joue un rôle majeur. Dans l’estomac, le milieu est très acide. Cela favorise l’absorption des médicaments qui sont des acides faibles. Dans l’intestin grêle, le pH est plus neutre ou basique, ce qui permet l’absorption de la majorité des médicaments, qu’ils soient acides ou bases faibles.
La vitesse de la vidange gastrique est aussi un facteur clé. Si votre estomac se vide lentement, le médicament mettra plus de temps à atteindre l’intestin, où l’absorption est la plus efficace. C’est pourquoi certains médicaments doivent se prendre à jeun et d’autres pendant les repas.
L’effet de premier passage et la notion de biodisponibilité
Lorsqu’on prend un médicament par voie orale, il ne passe pas directement dans la circulation générale. Après avoir traversé la paroi de l’intestin, il est d’abord dirigé vers le foie par la veine porte. C’est ce qu’on appelle l’effet de premier passage hépatique.
Le foie est une usine de nettoyage. Il va immédiatement métaboliser (transformer ou inactiver) une partie du médicament avant même qu’il ait eu la chance d’agir. Cet effet de premier passage réduit la quantité de principe actif qui atteint réellement la circulation sanguine.
Qu’est-ce que la biodisponibilité ?
La biodisponibilité (notée F) est le pourcentage de la dose administrée qui atteint vraiment la circulation sanguine sous forme inchangée. Une biodisponibilité de 70 % signifie que sur 100 mg de médicament avalé, seuls 70 mg sont disponibles pour agir dans le corps.
La voie d’administration influence directement cet effet. Par exemple, une administration par injection intraveineuse (IV) a une biodisponibilité de 100 %. Le médicament est directement dans le sang, il n’y a pas de premier passage.
Certaines voies permettent d’éviter ou de limiter ce phénomène :
- La voie sublinguale (sous la langue) : le médicament passe directement dans les capillaires sanguins, sans passer par le foie.
- La voie transdermique (patch) : l’absorption se fait à travers la peau, directement dans la circulation.
- La voie rectale : elle évite en partie le passage par le foie.
Phase 2 : La Distribution (D) du principe actif dans l’organisme
Une fois dans le sang, l’étape de distribution commence. Le médicament ne reste pas dans les vaisseaux sanguins ; il voyage pour se répartir dans les différents tissus et organes du corps : muscles, graisse, cerveau, etc.
Cette répartition n’est pas uniforme. Certains médicaments ont une affinité pour certains tissus et vont s’y concentrer. La distribution détermine où le médicament va agir et à quelle concentration.
Liaison aux protéines plasmatiques et forme active
Dans le sang, de nombreux médicaments ne circulent pas librement. Ils se lient de manière réversible à des protéines présentes dans le plasma, comme l’albumine.
Il faut imaginer ces protéines comme des taxis. Le médicament (le passager) monte dans le taxi et se laisse transporter. Tant qu’il est dans le taxi, il ne peut rien faire. Il est inactif. Cette partie du médicament est la forme liée.
- La forme libre est la seule qui est pharmacologiquement active. C’est la seule qui peut quitter les vaisseaux sanguins pour atteindre les tissus et exercer son effet.
- La forme liée sert de réservoir. Au fur et à mesure que la forme libre est éliminée, la forme liée se détache des protéines pour maintenir un équilibre.
Le pourcentage de liaison varie beaucoup d’un médicament à l’autre :
- Forte liaison (> 90 %) : dicoumarol, phénytoïne. Une très petite fraction du médicament est active à un instant T.
- Liaison moyenne : phénobarbital, théophylline.
- Faible liaison (< 10 %) : digoxine, paracétamol. La majorité du médicament est sous forme libre et active.
Volume de distribution et accumulation tissulaire
Le volume de distribution (Vd) est un paramètre pharmacocinétique un peu abstrait. Il ne correspond pas à un volume physiologique réel. C’est un volume théorique qui décrit l’ampleur de la distribution d’un médicament dans le corps.
Un Vd élevé signifie que le médicament quitte massivement le sang pour se concentrer dans les tissus (graisse, muscles). Sa concentration plasmatique sera faible. À l’inverse, un Vd faible indique que le médicament reste principalement dans la circulation sanguine.
Exemple de volume de distribution :
Un médicament avec un Vd de 5 L (le volume sanguin) reste dans le sang. Un médicament avec un Vd de 500 L est très largement distribué et accumulé dans les tissus, bien au-delà du volume total du corps humain.
Certaines zones du corps sont plus difficiles à atteindre, comme le cerveau. Il est protégé par la barrière hémato-encéphalique, une sorte de filtre très sélectif qui empêche de nombreuses substances de passer. Seuls les médicaments très liposolubles ou utilisant des transporteurs spécifiques peuvent la franchir.
Phase 3 : Le Métabolisme (M) ou la biotransformation
Le métabolisme est l’ensemble des transformations chimiques que le corps fait subir au médicament. L’objectif principal est simple : transformer une substance liposoluble en une substance plus hydrosoluble (soluble dans l’eau).
Pourquoi ? Car les reins, principal organe d’élimination, ne peuvent évacuer efficacement que les composés hydrosolubles. Le métabolisme est donc une étape de préparation à l’excrétion.
L’organe central du métabolisme est le foie. Il contient une grande variété d’enzymes, notamment la famille des cytochromes P450, qui sont les principaux acteurs de ces transformations. Les produits issus de cette transformation sont appelés métabolites.
Un métabolite peut avoir plusieurs destins :
- Être inactif : C’est le cas le plus fréquent. Le médicament est « désactivé » par le foie.
- Être actif : Parfois, le métabolite a une activité pharmacologique propre, parfois même plus puissante que celle du médicament de départ.
- Être toxique : Dans certains cas, le métabolisme peut générer des composés nocifs pour l’organisme. C’est le cas du paracétamol à haute dose.
Le concept de pro-drogue
Une pro-drogue est un médicament administré sous une forme inactive. Il doit obligatoirement subir une transformation métabolique dans le corps pour devenir un composé actif. C’est une stratégie pharmaceutique qui peut améliorer l’absorption d’un médicament ou cibler son action.
Par exemple, la capécitabine (Xeloda), utilisée en chimiothérapie, est une pro-drogue. Elle est convertie en sa forme active, le 5-fluorouracile, préférentiellement au niveau des tissus tumoraux. Cela permet de concentrer l’effet toxique sur les cellules cancéreuses et de limiter les effets secondaires.
Phase 4 : L’Élimination (E) ou l’excrétion du médicament
La dernière étape du parcours est l’élimination. Le corps se débarrasse du médicament (sous sa forme initiale ou sous forme de métabolites) pour nettoyer l’organisme. C’est la phase de métabolisme élimination.
Plusieurs voies sont possibles pour cette « sortie », même si deux d’entre elles sont largement majoritaires.
Les voies d’élimination
L’organisme dispose de plusieurs portes de sortie pour les médicaments et leurs métabolites :
- La voie rénale (urine) : C’est la voie d’élimination la plus importante pour la plupart des médicaments et surtout pour leurs métabolites hydrosolubles. Le rein filtre le sang, et les substances sont évacuées dans l’urine.
- La voie biliaire/hépatique (fèces) : Le foie peut aussi excréter directement des médicaments dans la bile. La bile est ensuite déversée dans l’intestin, et les substances sont éliminées dans les selles.
- Les autres voies : Elles sont minoritaires mais existent. L’élimination peut se faire par les poumons (anesthésiques gazeux), la sueur, la salive ou même le lait maternel.
La notion de clairance
La clairance (CL) est le principal paramètre pharmacocinétique qui quantifie l’élimination. Elle représente le volume de sang (ou de plasma) totalement épuré du médicament par unité de temps. Elle s’exprime généralement en ml/min ou L/h.
Il ne faut pas la confondre avec la quantité de médicament éliminée. La clairance est un débit d’épuration. Une clairance élevée signifie que le corps est très efficace pour nettoyer le sang de ce médicament.
La clairance totale est la somme des clairances de chaque organe impliqué dans l’élimination (rénale, hépatique, etc.).
Clairance totale = Clairance rénale + Clairance hépatique + ...
La formule de la clairance d’un organe est : CL_organe = Q * (C_A - C_V) / C_A où Q est le débit sanguin de l’organe, C_A la concentration à l’entrée et C_V la concentration à la sortie.
Paramètres clés : la relation concentration-effet et l’index thérapeutique
L’objectif de toute administration de médicament est d’obtenir une concentration plasmatique suffisante pour être efficace, mais sans atteindre un niveau qui deviendrait toxique. C’est tout l’enjeu de la posologie.
La relation concentration-effet (ou PK/PD) permet de définir une « fenêtre » dans laquelle le traitement est optimal. Cette fenêtre est appelée l’intervalle thérapeutique ou la marge thérapeutique.
Cet intervalle est délimité par deux seuils de concentration :
- Le seuil thérapeutique : C’est la concentration minimale en dessous de laquelle le médicament n’a pas ou peu d’effet.
- Le seuil de toxicité : C’est la concentration maximale au-delà de laquelle les effets indésirables ou toxiques commencent à apparaître.
L’écart entre ces deux seuils définit l’index thérapeutique. Si cet index est élevé, le médicament est relativement sûr : un petit surdosage a peu de chances d’être dangereux. Mais si l’index est faible, la manipulation du médicament est délicate.
Certains médicaments ont un index thérapeutique faible, ce qui exige une surveillance très stricte de leur concentration dans le sang :
- Lithium
- Phénytoïne
- Digoxine
- Théophylline
Pour ces médicaments, la dose doit être ajustée très précisément pour chaque patient afin de rester dans la bonne fenêtre, garantissant à la fois l’effet et la sécurité.
L’ensemble de ces études pharmacocinétiques est crucial. Elles permettent de déterminer la meilleure voie d’administration, la bonne dose et la fréquence des prises. Comprendre le devenir du médicament dans l’organisme, c’est la clé pour utiliser les médicaments de façon plus efficace et plus sûre. Les données obtenues sont essentielles pour le développement pharmaceutique et la pratique clinique quotidienne.



